
使用Biopython从gff文件提取gene的位置以及gene的id,再从fna文件提取gene序列
使用Biopython从gff文件提取gene的位置以及gene的id,再从fna文件提取gene序列
·
使用Biopython从gff文件提取gene的位置以及gene的id,再从fna文件提取gene序列
需要注意的是 Biopython存储到parse里面的并不是和文件的 染色体的顺序一致,在这里对字典嵌套列表,保存基因的位置以及其他信息,biopython读取的序列切片是从0开始的,这里它读取的基因的位置整体偏移1位

头部几个和NcBI里面的对比一下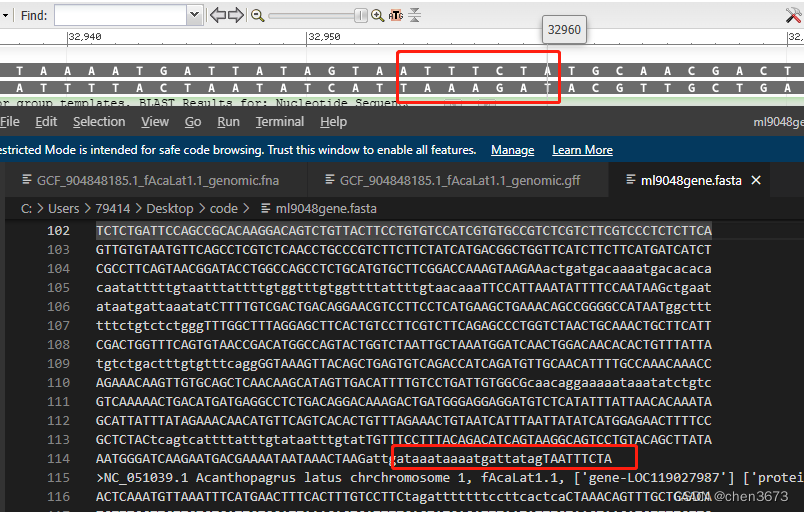
对比一下尾部几个碱基
from Bio import SeqIO
from BCBio import GFF
import re
def get_geneLocation_geneId(in_file,gene_len):
"""
本函数主要应用于从gff文件提取gene的location以及gene的id
in_file 为 输入的 gff文件
gene_len 为 需要提取的基因长度
"""
in_handle = open(in_file)
gene_information =dict()
for rec in GFF.parse(in_handle):
j=0
i=0
while j<len(rec.features):
if rec.features[j].type=='region':
tem = str(rec.features[j].id.split(':')[0])
gene_information[tem]=[]
if rec.features[j].type=='gene':
gene_location_t=[int(s) for s in re.findall(r'-?\d+\.?\d*', str(rec.features[j].location))]
if gene_location_t[1]-gene_location_t[0] < gene_len :
gene_information[tem].append([gene_location_t[0],gene_location_t[1],\
str(rec.features[j].qualifiers['ID']),\
str(rec.features[j].qualifiers['gene_biotype']),\
str(rec.features[j].location)])
i+=1
j+=1
in_handle.close()
return gene_information
def get_gene_gff_fna(file_path_gff,file_path_fna,save_path='./',gene_len=10000):#"GCF_904848185.1_fAcaLat1.1_genomic.gbff"
'''
本函数主要是用来从fna文件里面提取gene序列的
'''
return_gene=[]
gene_information= get_geneLocation_geneId(file_path_gff,gene_len)
with open(save_path+'.fasta','w',encoding='utf-8') as f:
for seq_record in SeqIO.parse(file_path_fna, "fasta"):
tem = str(seq_record.description.split()[0])
for j in range(len(gene_information[tem])):
return_gene.append(str(seq_record.seq[gene_information[tem][j]
下面的代码是添加的文件操作,把提取到的gene保存到*.fasta文件里面,,,具体的保存格式上图对比图有
from Bio import SeqIO
from BCBio import GFF
import re
import os
def get_geneLocation_geneId(in_file,gene_len):
"""
本函数主要应用于从gff文件提取gene的location以及gene的id
in_file 为 输入的 gff文件
gene_len 为 需要提取的基因长度
"""
in_handle = open(in_file)
gene_information =dict()
for rec in GFF.parse(in_handle):
j=0
i=0
while j<len(rec.features):
if rec.features[j].type=='region':
tem = str(rec.features[j].id.split(':')[0])
gene_information[tem]=[]
if rec.features[j].type=='gene':
gene_location_t=[int(s) for s in re.findall(r'-?\d+\.?\d*', str(rec.features[j].location))]
if gene_location_t[1]-gene_location_t[0] < gene_len :
gene_information[tem].append([gene_location_t[0],gene_location_t[1],\
str(rec.features[j].qualifiers['ID']),\
str(rec.features[j].qualifiers['gene_biotype']),\
str(rec.features[j].location)])
i+=1
j+=1
in_handle.close()
return gene_information
def get_gene_gff_fna(file_path_gff,file_path_fna,save_path='./',gene_len=10000):#"GCF_904848185.1_fAcaLat1.1_genomic.gbff"
'''
本函数主要是用来从fna文件里面提取gene序列的
'''
return_gene=[]
gene_information= get_geneLocation_geneId(file_path_gff,gene_len)
i=0
with open(save_path+'.fasta','w',encoding='utf-8') as f:
c=0
ge=0
for seq_record in SeqIO.parse(file_path_fna, "fasta"):
tem = str(seq_record.description.split()[0])
for j in range(len(gene_information[tem])):
f.write('>')
f.write(seq_record.description.split()[0])
f.write(' ')
f.write(str(seq_record.description.split()[1]))
f.write(' ')
f.write(str(seq_record.description.split()[2]))
f.write(' ')
f.write('chr')
for aa in seq_record.description.split()[5:-4]:
f.write(str(aa))
f.write(' ')
f.write(gene_information[tem][j][2])
f.write(' ')
f.write(gene_information[tem][j][3])
f.write(' ')
f.write(gene_information[tem][j][4])
f.write('\n')
return_gene.append(str(seq_record.seq[gene_information[tem][j][0]:gene_information[tem][j][1]]))
n=0
for m in return_gene[-1]:
if n<80:
f.write(m)
n+=1
else:
f.write('\n')
n=0
f.write('\n')
c+=1
print(c)
i+=1
print('extra gene over')
return return_gene,gene_information

技术共进,成长同行——讯飞AI开发者社区
更多推荐

 2
2 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)